We will help you with dissertation
By providing extensive dissertation writing service
Just click and the writing will begin
Trusted by 4370 satisfied students

Academic Writers
Locate an online academic writer you will be excited to work with. Each maven was thoroughly hand-picked and is cunning in their field. Get yourself familiar with profiles, scores, customers’ reactions to hire an elite paper writer who ticks all boxes.
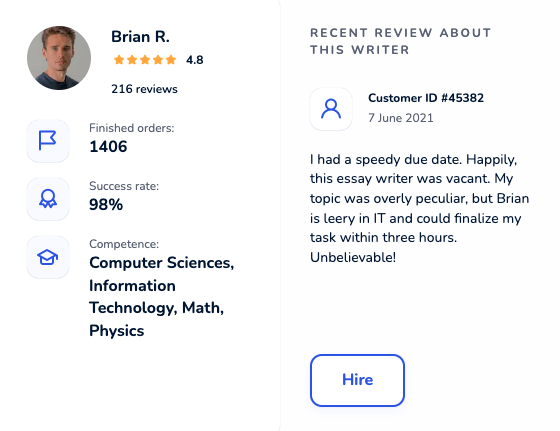
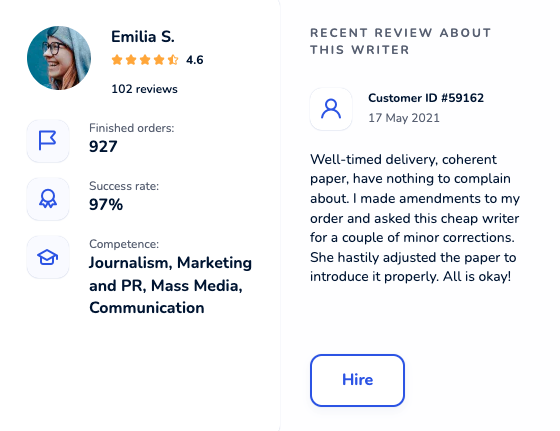
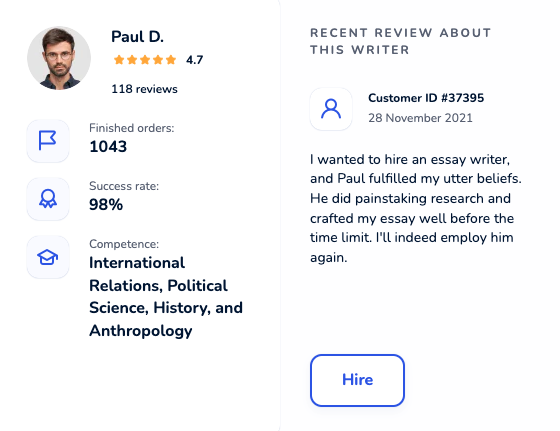
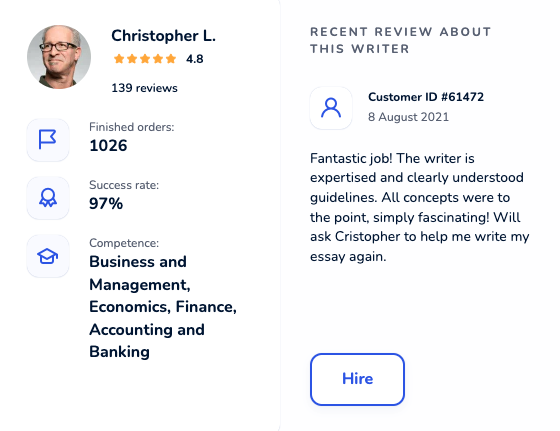
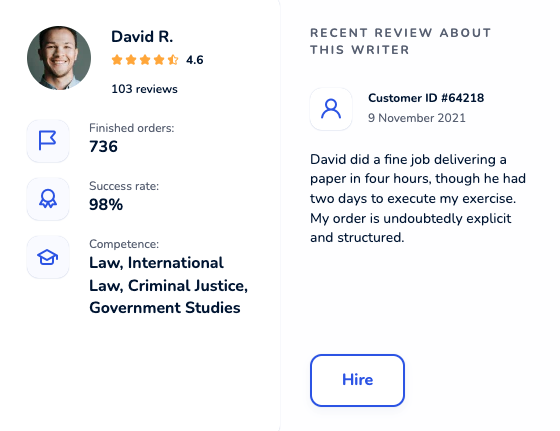
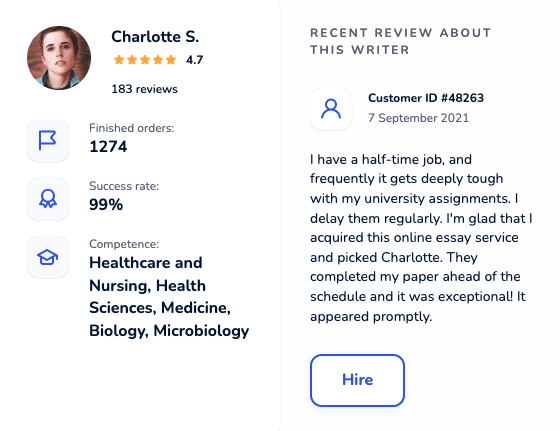

Dissertation Writing Service Packed With Benefits
While being a client-oriented dissertation writing service is among our service assets, purchasers can’t help but appreciate other handy features. Discover perks, embarking on the adventure to academic triumph.
100% Uniqueness
Get original papers customized to your needs as you pay someone to write your dissertations. Each work is meticulously scanned for plagiarism and duplicates. Request an originality report if needed.
Ultimate confidentiality
Customers' private data is a sheer priority. Our academic writing service neither collects nor reveals your identity, discloses personal data, or passes sensitive data to third parties.
Timely turnaround
Never miss a shortest deadline with fast dissertation writing service help. Our experts possess strong time-management skills, consistently delivering papers timeously.
Small corrections
Count on rigorous compliance as you pay for dissertation writing. Request free unlimited revisions in the opposite event. You have 14 days from the deadline to ask for rectifications.
Paper Writing Service Doing Any Assignment With Quality in Mind
Cooperating with experienced specialists gives immediate access to all paper writing services. Get acquainted with varied assignments paper writers present and inspect samples to see the quality we offer.
- Term Paper
- Lab Report
- Admission Essay
- Case Study
- Research Paper
- Book Report
- Scholarship Essay
- Peer Review
- Capstone Project
- Movie Review
- Personal Statement
- Literature Review
- Thesis/Dissertation
- Statistics Report
- Critical Thinking
- Annotated Bibliography
- Coursework
- Business Plan
- Miscellaneous
- Math Assignment
- History Assignment
- Business Assignment
- Science Assignment
- Physics Assignment
- Biology Assignment
- Marketing Assignment
- Computer Sciences
- Chemistry Assignment
- Nursing Assignment
- Economics Assignment
- Engineering Assignment
- Statistics Assignment
- Psychology Assignment
- Finance Assignment

Testimonials About Our Dissertation Writing Services
Our customer reviews about speak for themselves. Check what students think about our best dissertation writing services. Choose the right filters for easier feedback navigation.






Estimate How Much It Costs to Write My Dissertation
The total sum hinges on writing service, due date, general complexity, and personal preferences. Calculate the definitive price and choose from flexible options to pay for dissertations.


Get the Best of Our Dissertation Service
The sky’s the limit regarding the advantages of academic dissertation writing service. Whether you’re suffering from unbearable pressure or have a crucial demand, we will cheerfully devise a solution.
- Get rid of writing assignments and piles of homework.
- Receive high-quality dissertations tailored precisely to your needs.
- Spend time on things that matter most while we are dealing with paperwork.
- Get rid of writing assignments and piles of homework.
- Receive high-quality dissertations tailored precisely to your needs.
- Spend time on things that matter most while we are dealing with paperwork.
EASE YOUR LIFE:
Getting professional dissertation writing help online has not once been easier! Give us your requirements, and top-ranked assignment writers will handle the rest.
Students Also Ask About Our Dissertation Writing Services
Everything you wish to know about us
Positively! Our service is a legitimate paper writing service proffering full-scale writing assistance. Subscribe and use an enjoyable cheap reliable dissertation writing service trusted by undergraduates worldwide. Binding regulations oversee steadfast teamwork. We strictly obey the classy terms and policies stated on the site, securely storing personal information. You get a custom chunk to utilize for quotations or further analysis by trusting a legit dissertation writing service.
At our service, you can get someone to write your dissertation for you quickly. The timeframe required to write paper for you varies hinging on service kind and volume. More difficult duties may involve an exceedingly thorough examination. Analyze minimum periods required to yield professional writing service online:
3 hours – High school, college, university
12 hours – Masters and Doctorate.
Delineate accurate guidelines hire an academic writer to help. We will speedily perfect your undertakings. Reminder: Our service proposes lower pricing for orders with outstretched time cutoffs. Greater space for exploration enables decreased expenditures.
We treat respective inquiries with significant involvement, tying you with a prime online paper writer. We cherry-pick top academic writers during an intense screening act to attain this objective. Contestants should hold either Master’s or Ph.D. degrees, bear powerful writing knacks, and have verified sturdy expertise. Most intelligent and polymathic scholars join our crew. “Write a paper for me” queries are in neat hands.
We foster diversity and hire professional writers competent in assorted domains. Be it Sociology or Jurisprudence. You will definitely spot the best academic writer. If you have difficulty choosing a paper writer, contact our Support Center. Committed agents will be increasingly blissful to uncover a properly aligned match based on your directions.
Our agency suggests agile payment methods to pay for dissertations online smoothly. We presently accept Visa, Mastercard, UnionPay, American Express, Discover, JCB, Google Pay, Apple Pay. Our plethora of payment systems stems from a desire to warrant healthy flow and ultra anonymity. Your transaction will be processed per a gateway defended by cutting-edge technology. Residue in confidence – your data is safeguarded, whereas you are paying someone to write your dissertation.
Our writing service is a website that writes dissertations for you with high intellectual rules in mind. Our paper writers scrupulously explore precise topics, creating an original fraction based on prior research and customer requirements. Our masters double-check upshots for uniqueness prior to submitting. Sequentially, rigid quality control and internal databases give authentic results.
We constantly verify execution and organize systematic tests. Connoisseurs are accustomed to scholarly updates and can cater marvelous help with writing an dissertation.
Our dissertation writing help service specializes in over 30 wide academic ranges and sundry interdisciplinary courses. Some faculties are divided into dozens of derivative branches and are not incorporated into our menu. Pinpoint “Other” if you can’t distinguish your specialization in a drop-down list.
We extremely seriously urge specifying a craved department in the “Paper Details” field. While messaging, “I need help writing an dissertation”, enumerate exhaustive instructions. This will allow us to assign the most seasoned writer.
To reserve an examiner, we require that you pay to write dissertation online upfront. This way, paper writers vouch that you are interested in cooperation and will succeed in writing an online assignment beyond expectations. Consecutively, we pledge the finest grade.
Get Dissertation Help in 3 Simple Steps
This can be the beginning of a fruitful partnership.
Follow these stages to get paper help from an expert.
Share dissertation writing requirements
Fill in the order form with aggregated paper instructions and attach supplementary files. Perpetuate utmost accuracy so that our paper expert can kick-start your order.
Meet a paper writer
Browse through guru bios to obtain the most suitable writer. To indemnify our specialists, you will need to pay for dissertation online sooner.
Get your dissertation written for you
Once the writing assignment is good to go, you will be able to evaluate it. If satisfied, download the finished paper immediately, and don't forget to leave a testimonial.
Professional Dissertation Writing Service Where Quality Meets Efficiency
Our service presents leading professional dissertation writing services online. We leverage skillful academic dissertation writers online to accommodate sophisticated solutions. Our world-class team is dedicated towards transforming your scholastic journey, bringing better results.
Dissertation Writing Service Online - The Best Solution for Every Ph.D. Candidate
Dissertation writing is a stressful and challenging task. Few candidates can handle it without any additional help. Most students eventually come to a point where they want to give up on their thesis, and that’s when our professional PhDify.com team comes in to give them help and support. We provide PhD dissertation writing service. Every student in need of support will find it here.
With our platform you get numerous benefits, including a wide selection of unique topics, fresh ideas, authentic information for your work, timely responses from our writers, safety of your personal information, etc. By using our custom dissertation writing services, scholars can be confident that talented and experienced writers will assist them with both conducting research and writing their paper. We treat our clients with the utmost respect and take special care to value your time and meet your exact needs.
PhDify has been in operation since 2004. We have a proven reputation of punctuality, quality, and reliability. No other dissertation service offers the kind of guarantees we do: on-time delivery, unlimited revisions, and a 100% money-back guarantee.
Our Guarantees
100% Original Papers
Zero-Tolerance Plagiarism Policy
In-Depth research
On-Time Delivery
Constant Contact with Your Writer
Say "Write My Paper" and Get Fitting Dissertation Help Online
Expect especially punctual help with dissertation writing when you order papers writers at our writing service. Time is priceless legacy juniors own. Hardly anyone can afford to waste precious instances anticipating custom dissertation help service. We value on-time delivery. Attentive papers writing service works around-the-clock to prevent shoppers from biding for ages.
We recruit writers who manage effective planning. Precision, promptness, and devotion define dissertations help online. When you order “write paper for me”, be sure that it will arrive swiftly. Our team of professional writers strives to wrap up a final draft sufficiently before the due date, whether you lust it harshly or in 3 weeks.
Hire a paper writer for dear tasks. Usually, it takes 3 hours to finish a paper for high school, college, or Bachelor students. The duration necessary for postgraduate assignments reaches 12 hours. However, we would be grateful for having elongated target dates for tasks implying serious study.
Dissertation Services Delivering Plagiarism-Free Writing Help
The founders of our writing dissertation online service are former classmen. We comprehend that plagiarism can cost clients their solid position. When ordering professional writing services, be certain that the content contains no signs of plagiarization. Unfortunately, not all websites that write dissertation for you function responsibly. Some companies descend to ‘copy-paste’ acts jeopardizing your image. StudyCrumb’s paper writing help eliminates piracy risk. All orders are validated through innovative originality software. Here’s how we achieve this aim in a nutshell:
Freelance writers carefully read clients’ instructions and enforce all-inclusive topic research.
Unique ideas are developed contingent on preliminary investigation.
Academic writers online cite sources and run plagiarism monitoring.
Particular paper passes an internal plagiarism detection scheme.
You receive an original cheap paper written exclusively for you.
“Can you write my dissertation for me without plagiarism”, you might marvel. We evidently can. If you doubt the service’s worth, feel liable to claim an originality report. Elect this premium innovation while placing your paper writing order, or contact Customer Support.
Cheap Dissertation Writing Service with Affordable Prices
Visiting our platform, chances are you need cheap papers. Nobody wants to outlay a fortune on websites that write papers for you. We charge reasonably so that you and your dissertation writer for hire are satisfied. Tariffs differ contingent on the professional writer service category. They begin with $6 for editing and $15 for original papers. Unlike other dissertation writing websites estimating prices for a 275-word page, at StudyCrumb, you get a 300-word text.
If you want to profit from the cheapest write my dissertation help, there is an elementary trick. Economize by allotting prolonged deadlines for fulfillment. To shake things up, StudyCrumb has great privileges that will surely win your heart:
Referral program: get $30 for each referred friend
Loyalty procedure: enjoy generous discounts for standing by StudyCrumb.
Holiday deals: use amazing cut-rate on anniversaries and selective occasions.
Assure to periodically revise cheap dissertation writer service for upgrades, never to overlook deals.
Write My Dissertation For Me Cheap Without Impacting Quality
Some users feel reluctant about paying someone to write an dissertation. They haunt services that “write my papers for cheap” to cut disbursements. Others suspect fraud and avoid being deceived. Seekers pay someone to write dissertation substantially less to justify coordination. Commonly, superfluous vigilance brings about adverse effects. Bidders are eventually dissatisfied with mediocre output. So, where do I retrieve dependable and cheap write my dissertation services? At Studycrumb.com!
In our book, “cheap dissertation services” means “immaculate reasonably-priced assistance”. We managed to furnish budget-friendly commissions without compromising fineness. Our service is a sharp formula of brilliance and affordability. If you are questioning, “can someone write my paper for me cheap and well”, the remedy is apparent.
How to Outsource Help from Academic Writer Online
Are you looking for a service to expose paper writers for hire? You’ve come to the honorable website for dissertations! Here’s a piecemeal manual to a rewarding collaboration.
Register an account.
Particularize your email address and phone number for smooth clarifications or reporting. Ascertain our correspondence doesn't fall into spam lists. Create a passcode with digits, letters, and symbols in the settings.
Describe essentials.
Enclose pivotal circumscribing and templates. At our service, you can engage a top dissertation writer in your specific sphere. We welcome variety, going to great lengths to gather talented specialists.
Skim through portfolios.
Familiarize yourself with each enthusiast's ample background and maintain a direct conversation. Consider these things when choosing specialists: Occupation Satisfaction rate Past orders Recent evaluations.
Salute your author.
All writers can help to write my dissertations for me in line with the requirements. Connect with experts, and figure out comprehensive info and their abilities.

Write My Dissertation For Me! Other Details About Writing Service
Have questions about our website for dissertation? Surf through answers downwards to recognize an instantaneous resolution.
We are artists congenially operating on the market and assisting learners like you. We coordinate integral processes based on stringent policies attest to top-notch products and privacy. No personal data will be transferred to unauthorized bystanders after you pay someone to write an dissertation at our service.
When undergrads choose our website that writes papers for you, we deliver high-quality works with consistent formatting. We administer most referencing styles, including but not restricted to APA, MLA, Chicago, Turabian, etc. Indicate the desired format in the order form, so we complete your paper accordingly.
Our developers designed a greatly intuitive site that writes essays for you. As you are ready to hire the best helper attainable at our online paper writing service, navigate to the upper catalog and click the “Order Now” button. You will be redirected to the order form. Fill out the required fields with specifications, and upload additional materials. Our essay writer website has improved easy-to-use integrated premium additions. Add auxiliary specialties to help essay writing service order. Procure a phone number, valid email address, and password to receive further counseling.
Pricing for academic dissertation writing services is conditional on four factors:
Type of academic paper
Number of pages
Academic level
Deadline.
Personal precedency additionally determines the end price. If you want to pay someone to do your dissertation and benefit from enhanced comfort, embrace premium add-ons. From “Premium writer” to “Extra quality checkup”, we serve the best dissertation writing for brilliant results.
Go no further if you are looking for cheap writers online. We believe that students shouldn’t melt their entire savings on academic consultancy. Our service nourishes cost-wise surroundings for customers. We respond to each “write my paper cheap” call with an affordable dissertation writing service online. Obliterate unreasonably pricey rates and hidden expenses.
First and foremost, as a reputable paper writer website, we exert ourselves to adorn unusual commodities. Every patron is preserved by policies that regulate future activity. They confirm user protection and consistency of. Though bad quality is uncommon for us, you are entitled to free alteration or compensation. We encourage you to award detailed recalls on a mismatch between the primary requirement and the eventual draft. Our Quality Assurance Team will closely scrutinize your context per the protocol, resolving your issues complying with Money-Back Guarantee.
The user-friendly interface of our essay site generates tracking functionality through a customer dashboard. Audit your order progression and chat with the writer directly to formulate conditions. As you form a requisition “write my paper for cheap”, your order will be assigned ‘Unpaid’ status. Once you pay, the order will move into the “Paid” state. As your creator takes on a task, the status will be altered to ‘In progress’. A specialist may contact you to discuss specifics. Once your paper is done, you will be notified via mailbox notification.
When sophomores hire someone to write an dissertation online at our writing service, we promise an uneasy judgment. It might be hard to set your sights on a sole good academic writer with this abundant selection. Itemize specs – our assistants will induce a pertaining reservation. Feel free to approach our Customer Support. Consultants will release the best person.
Do My Dissertation Urgently!
Whether you are dealing with high-school assignments or thirsty for doctoral assistance, you can always count on do my dissertation paper writing service. No matter what academic level you prefer or how intricate a task is, specialists are trained enough to supply the best dissertation services.
Pay for Dissertation and Stay Confident
We promote secrecy. An entity can pay someone to write my dissertation for cheap, knowing that the details are safe and sound. Once an order is placed, it is required to pay someone to do your dissertations. We permit multiple payment options to single out the most convenient manner to ship funds. Our dissertations writing services currently accept most major bank cards and digital wallets for imperative payment.
All payment mechanisms are completely decent. Stipulations entered will be protected by the safety measures, s.a. latest encryption technology and won’t be visible to anyone. When paying for someone to write an dissertation, you foresee that your personality will reside anonymously. At our writing service, we certify that no personal information will be passed to secondary parties. Help in dissertation writing will remain confidential unless you decide to split customer experiences yourself.
Find Someone to Write My Paper Order on Any Discipline
Googling “hire someone to write a paper for you” or “cheap writing paper service”, you will unfold manifold dissertation websites displaying help. Most writing dissertation services brag about superiority. But not all academic writer websites can sustain this top-class service and cover variant disciplines.
We reckon that the best dissertation service should be accomplished in diverse topics. Our team consists solely of professional dissertation writers online who victoriously graduated from leading universities. They have a splendid level of preparation and are knowledgeable in various themes.
Whether you need help with Humanities, Social, Natural, Formal, or Applied Sciences, you will categorically perceive a paper expert adept in your region. Visual Arts, Linguistics, Religion, Theology, Geography, Algebra, Geometry, Public Administration, Economy, Sports, Military, and Architecture are just a few erudite areas. Writers nail papers on bullying, cybercrime, capital punishment, the death penalty, harassment, domestic violence, police brutality, abortion, marijuana legalization, gender discrimination, video games, vaccination, disorders, and diseases.
Write My Dissertations Online with the Best Paper Writers
There is bountiful usefulness to fancy appealing to pay people to write dissertations for me online. Hiring the best writers online lets students adhere to models. Our specialists know all ins and outs of custom writings and build impeccable outcomes.
Hiring the best assignment writers might be a heavy determination. Graduates repeatedly feel stuck hustling to pick out executors. You should acknowledge every aspect of your assignment. Luckily, super helpful educational writers at our service have a proven track record in this exigent industry. Driven by tremendous enthusiasm, they produce well-written papers with a systematized structure.
Pay for paper service from us and be prepared for the final bespoke work. Don’t hesitate to solicit an unpaid revision if you pine for minuscule fixes to adapt an assignment to your basic guidelines. Plus, you can use this variation several times. This particularity is available fourteen days from order expiration or the resulting draft. Although poor quality is not typical for us, we cultivated corporate warranties that give faith.
Need Urgent Dissertation Writing Help?
The upcoming submission date may force looking for academic companies that “write my dissertation today”. If a due date rears, but you haven’t initiated pursuing your paper, rely on writing dissertation help websites. You may have superfluous errands. Making a paper yourself isn’t an option. Our writing service is mature to dispense vital and cheap dissertation help. Handpick from hundreds of writers, give yourself room for maneuver and never skip a due date.
Keep in mind that some writers may have a high workload and be unable to kick off immediately. We highly advocate scheduling your assignments in advance, and setting extended deadlines. This way, your paper writing expert will have expanded time to investigate a matter’s scope. As a bonus, you will be able to save wider on “cheap write my paper” orders.
The choice is obvious. By imposing a “write me a paper” proposal, the applicant gets an ambitious backup for personal endeavors. Don’t be shy with prescriptions and documents, watching your cheap dissertation papers being governed.

Don't wait until too late – order
your paper now!
Entrust any task to experts, who will take good care of the ordered assignment.
Our Assistance Will Be Handy In:
Research process, data collection and analysis; Gathering sources and theoretical material; Consultancy on creating unique academic content; Editing your writing according to the highest standards; Formatting your papers and citing the sources in line with the latest requirements.
About Us
Contacts
Feel free to get in touch with us via phone or send us a message
- +1 585 282 63 46
- [email protected]
